苹果抗炭疽菌叶枯病基因的SSR标记筛选及遗传定位
苹果抗炭疽菌叶枯病基因的SSR标记筛选及遗传定位
培育抗病品种是一种经济有效的手段,成为解决苹果炭疽菌叶枯病的首选。传统的抗病育种主要依赖于植株的表现型选择 (P he-notypical selection),但是由于环境条件、基因间互作、基因型与环境互作等多种因素大大影响表型选择效率。如抗病性的鉴定就受发病的条件、植株生理状况、评价标准等条件的影响。一个优良抗病品种的培育往往需要花费7~8年甚至十几年时间。随着分子生物技术的快速发展,以DNA多态性为基础的分子标记技术以其表现稳定、数量多、多态性高等优点已被广泛的运用于植物遗传图谱的构建、控制重要农艺性状基因的标记遗传定位、种质资源的遗传多样性分析以及品种指纹图谱的绘制等方面,尤其是分子标记辅助选择 (molecular marker-assisted selection,MAS) 育种,相较传统育种能极大地提高育种的选择效率与育种预见性,受到人们的高度重视。
简单重复序列 (simple sequence repeats,简称 SSR) 又称微卫星(microsatellite) 广泛地分布于果树基因组的不同位置。SSR位点多态性的形成是基于基本单元重复次数的不同。由于每个SSR位点两侧一般都具有相对保守的单拷贝序列,所以可以根据此特点在SSR两侧序列设计一对特异引物来扩增 SSR 序列。通过对 PCR 产物进行聚丙烯酰胺凝胶电泳或琼脂糖凝胶电泳来显示不同 SSR 标记的分子多态性。由于SSR标记具有大量的等位差异、多态性好、操作简便、稳定等特点,已被广泛应用于作物的遗传图谱构建、指纹图谱绘制、目标性状基因的标记定位、物种起源进化及品种纯度鉴定等 (Hemmat,1994)。
本试验利用SSR标记与集团分离分析法BSA (bulk segregant anal-ysis) 相结合,快速有效地寻找与质量性状遗传的目标基因紧密连锁的SSR标记,用于分子标记辅助育种及抗病性的早期鉴定。
第一节 材料与方法
一、植物材料
本试验选择青岛农业大学苹果试验基地 (山东省胶州市) 2009年种植的,经过室内离体接种鉴定的 ‘金冠’ב富士’ 的207 株F
二、DNA的提取及检测
1.总DNA的提取
参考Doyle和Doyle (1987) 及 Cullings (1992) 提取基因组DNA的CTAB法,并加以改进 (附录一)。
2.DNA纯度及浓度的测定
(1) 利用1%琼脂糖凝胶电泳检测。取 4μl DNA 样品与 2μl 6×Lodding buffer 混匀,在 1%浓度的琼脂糖凝胶中电泳 (120V,30min),最后在紫外凝胶成像系统中成像并记录保存。
若成像为一条整齐、单一、清晰的 DNA 条带,且点样孔没有亮光,则表明所提样品较纯;若条带不清晰、拖尾或出现涂抹带,则表明 DNA 发生了降解,降解严重会看不到条带;若在胶片下部有弥散的荧光区出现,则表明样品中存有 RNA 杂质;若点样孔处有明显的亮光,则说明样品中含蛋白质和大分子杂质。
琼脂糖凝胶电泳检测方法见附录二。
(2) 分光光度计检测。运用分光光度计NanoDrop 2000 进行 DNA纯度及浓度的量化测定。
若 OD260/OD280 值在 1.8~2.0,并且 OD260/OD230 值大于2.0,则表示此样品DNA纯度适宜。
三、抗感DNA池的构建
将提取、纯化的基因组 DNA,稀释到浓度为10ng/μl。根据该组合群体的离体接种鉴定结果,将杂交后代单株分为抗病和感病两大类型。按照BSA分析方法的要求,选取 10 份高抗单株 (无任何病斑)的DNA,等量混合构建DNA抗池;选取10份高感单株 (病斑个数大于20) 的DNA等量混合构建DNA感池。两个基因池用于筛选与目标基因连锁的分子标记。
四、SSR分子标记的筛选与开发
1.SSR分子标记开发
从网站 https://www.rosaceae.org/gb/gbrowse/malus_x_domestica/下载目标区域的 contig 序列,然后通过网站 http://archive.gramene.org/db/markers/ssrtool搜索该区域碱基序列中所有的 SSR 位点。搜索参数设置为:碱基重复单位为 2、3、4、5、6 个碱基,相应的重复次数依次为 8 次、6 次、4 次、3 次、3 次。利用 Primer 3.0 P lus软件设计SSR引物,引物设计时应注意:引物与SSR位点间的距离一般大于50 bp 个碱基序列。引物 GC 含量为40%~70%,最适值为50%;引物长度在18~24 bp;退火温度50~65℃,左右引物退火温差小于 5℃;扩增产物片段大小在 150~350 bp。引物的评估利用Oligo软件进行,避免引物二聚体、发夹结构和错配等情况的发生。引物序列 (附表1)。所有引物由生工生物工程 (上海) 股份有限公司合成。
2.PCR扩增
SSR反应体系为15 μl,内含10 ng/μl基因组DNA 2 μl,1×Master Mix 7.5 μl,0.2 μmol/L左右引物各0.8 μl。进行初步筛选时的 PCR扩增程序为:94℃预变性 5min,然后按 94℃变性 30 s,55℃退火40 s,72℃延伸30 s的程序进行 10 个循环,每个循环的退火温度降
3.聚丙烯酰胺凝胶电泳
聚丙烯酰胺凝胶电泳的方法见附录三。
4.SSR标记的筛选
从 HiDRAS 网站 (http://www.hidras.unimi.it/) 和 GenBank (http://www.ncbi.nlm.nih.gov/genbank) 网站下载了 300 对均匀分布于苹果17条染色体上的已发表的 SSR 引物,在亲本及抗感池中进行初步筛选,选出在抗亲、抗池与感亲、感池中有多态性条带的引物,然后在207个做图群体上进行筛选。最终选出与抗性基因位点连锁的标记,根据所筛选出的SSR标记的已知信息,确定其所在的染色体,然后将 SSR 标记序列与苹果基因组数据库 (http://www.rosaceae.org) 进行BLAST比对,将其定位在染色体的具体位置上。初步定位后,从网站 https://www.rosaceae.org/gb/gbrowse/malus x domestica/下载与目标基因位点连锁的 SSR 标记间的 contigs序列,根据SSR标记设计的方法,设计了 276 对新引物。这些引物首先在抗亲、抗池与感亲、感池中进行筛选,将产生多态性条带的引物再进行群体验证。
对检测群体中各单株的 SSR 标记基因型分别赋值并记录,与抗池带型相同的记为 “A”,与感池带型相同的记为 “B”。将这些SSR标记在群体上的基因型数据进行孟德尔1R∶1S遗传符合度的卡方检验。并将表型抗性鉴定结果与标记基因型数据相结合,采用 JoinMap 4.0软件,对标记及抗性基因
5.SSR标记的序列分析
将筛选获得的与抗性基因
第二节 结果与分析
一、基因组DNA的检测
用CTAB法提取的苹果叶片基因组 DNA经1%的琼脂糖凝胶电泳检测,结果表明,DNA条带清晰,完整无降解 (附图3-1)。可以用于后续的研究。
二、抗性基因的分子标记筛选
从 HiDRAS 网站 (http://www.hidras.unimi.it/) 下载的 300 对均匀分布于苹果17条染色体上的已发表的 SSR 引物在亲本及抗感池中进行初步筛选,选出54 对在抗亲、抗池与感亲、感池中有多态性条带的引物。再将这54 对引物用于作图群体的207 个单株以筛选与抗性基因位点连锁的 DNA 标记。最终筛选出2 个可以清晰区分抗感双亲、抗感池和杂交群体抗感单株的 DNA 标记,CH01d08 和CH05g05。引物序列如表3-1中所示,因为这两个标记已被报道位于苹果15号连锁群上 (Liebhard et al.,2002),所以将苹果炭疽菌叶枯病抗性基因 (命名为
根据苹果基因组 CH01d08 和 CH05g05 标记之间的核苷酸序列,自行设计了276对SSR引物。按照上述方法进行筛选,最终筛选出9对引物能够扩增出清晰稳定的多态性条带的引物 (附图3-2、附图3-3),分别为 S0607039、S0607001、S0506206、S0506001、S0506078、S0405195、S0405127、S0304673、S0304011 (表3-1)。连锁分析表明,标记S0405127和S0304673与
SSR编号引物序列重复基序产物长度/bp退火温度/℃位点CH01d08aF:5′-CTCCGCCGCTATAACACTTC-3′R:5′-TACTCTGGAGGGTATGTCAAAG-3′ag29056MDC021953.346chr15∶13688903..13699651CH05g05aF:5′-ATGGGTATTTGCCATTCTTGC-3′R:5′-CCTGAAGCAAGGGAAGTCATAC-3′ag14356.5MDC016699.237chr15∶2343805..2349433S0607039F:5′-AACGCACCGACCCATTTC-3′R:5′-CCAGCTCGCATAACCACC-3′ct18654MDC011529.272chr15∶6103161..6122652S0607001F:5′-ATGAAAGCGAGTCGGAGTG-3′R:5′-GGGGAGGGTTGGTGGTTA-3′caggtcaggt26956MDC004171.329chr15∶5986277..6005012S0506206F:5’-GCTGAGATTTCCCCCATT-3′R:5′-GCTGCGGACACTGCTTAG-3′ttggatgtg24354MDC007696.347chr15∶5714203..5748693S0506078F:5’-AGAAAGGCCCTCAAACAG-3′R:5′-CTGCAGAAGGTGGGTATG-3′aaaagc30455MDC002692.183chr15∶5005415..5011924S0506001F:5′-CATGAAAAGGTAGGCAGTGG-3′R:5′-GAGGTTCTTGGGCAAGTGTT-3′acaaccaa30454MDC013564.245chr15∶5006247..5017709S0405195F:5′-AGACGGGCAAATTAGTTGAGAT-3′R:5′-TCCCTTCTATGATGAATGACACC-3′tg25853MDC016041.193chr15∶4672532..4691912S0405127F:5′-GGCACAATGTAGGAGGGATA-3′R:5′-GCTATGAGGAAATTGGCTCT-3′at33055MDC043871.6chr15∶4622388..4626535S0304673F:5′-GTTTGCACATTGTAATGCTG-3′R:5′-CAGTTTTCTAGTGATGTCGTTG-3′tg(ga)33353MDC013859.580chr15∶4121053..4135560
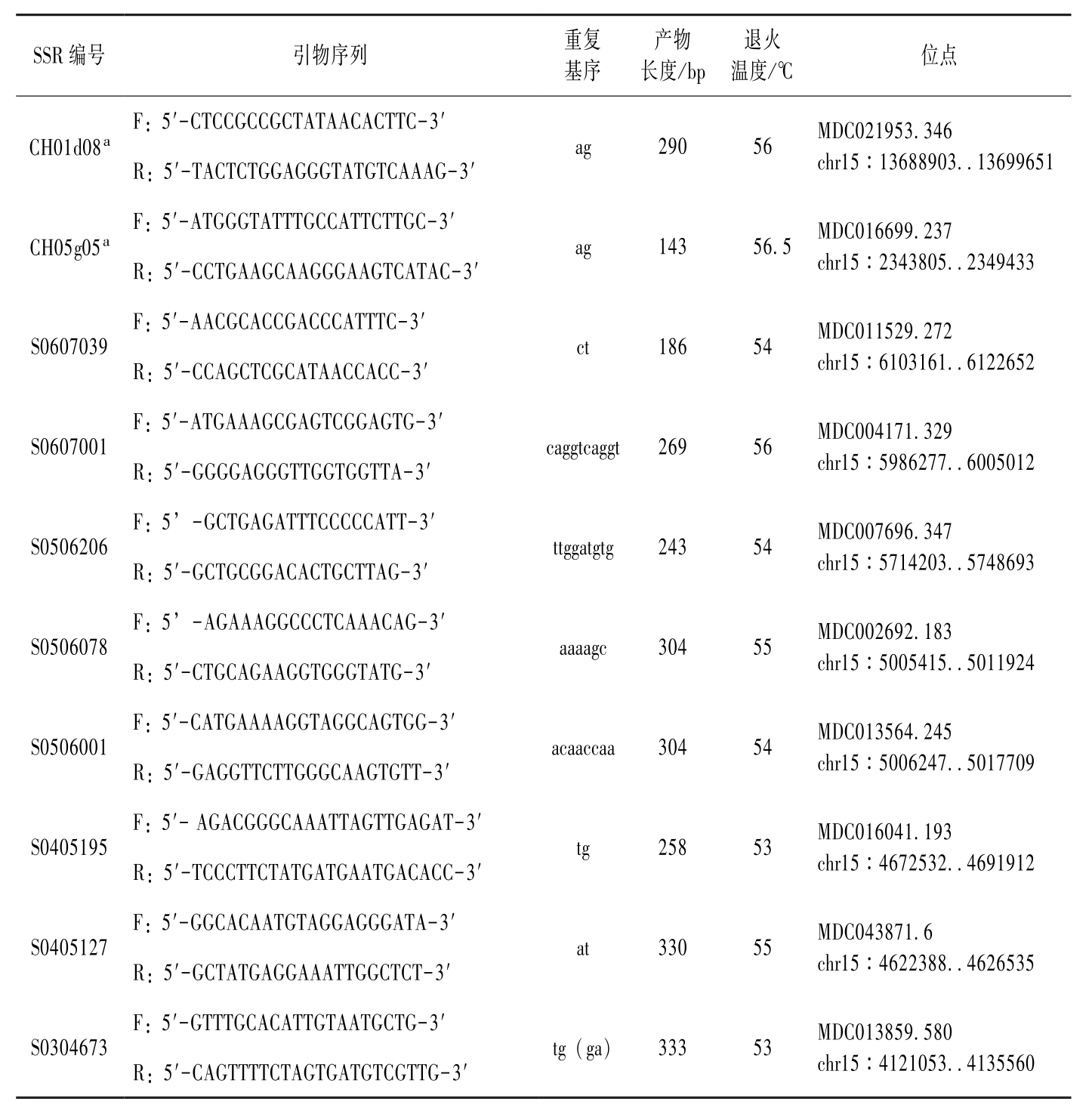
表3-1 定位在15号连锁群上与Rgls基因连锁的SSR标记序列及引物
SSR编号引物序列重复基序产物长度/bp退火温度/℃位点S0304011F:5′-GCCGAATCTGCGGAATTG-3′R:5′-TCCCACTTCCTCACCGTCTC-3′ag21056MDC015994.315chr15∶3183972..3196801
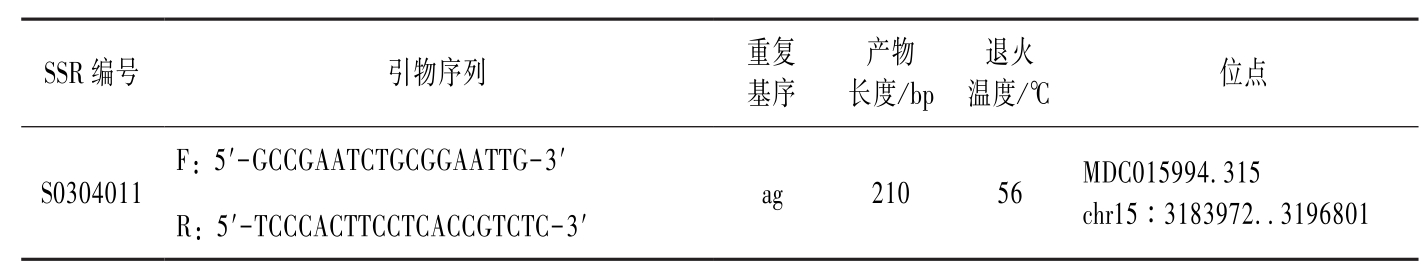
表3-1 定位在15号连锁群上与Rgls基因连锁的SSR标记序列及引物(续)-1
SSRmarkerObservedratio(R∶S)Expectedratio(R∶S)x2PS030401184∶123103.5∶103.53.670.06Ch05g0590∶117103.5∶103.51.760.18S030467393∶114103.5∶103.51.070.30S040512791∶116103.5∶103.51.510.22S040519588∶119103.5∶103.52.320.13S050607894∶113103.5∶103.50.870.35S050600192∶115103.5∶103.51.280.26S060700188∶119103.5∶103.52.320.13S060703998∶109103.5∶103.50.290.59S050620695∶112103.5∶103.50.70.40Ch01d08104∶103103.5∶103.500.96
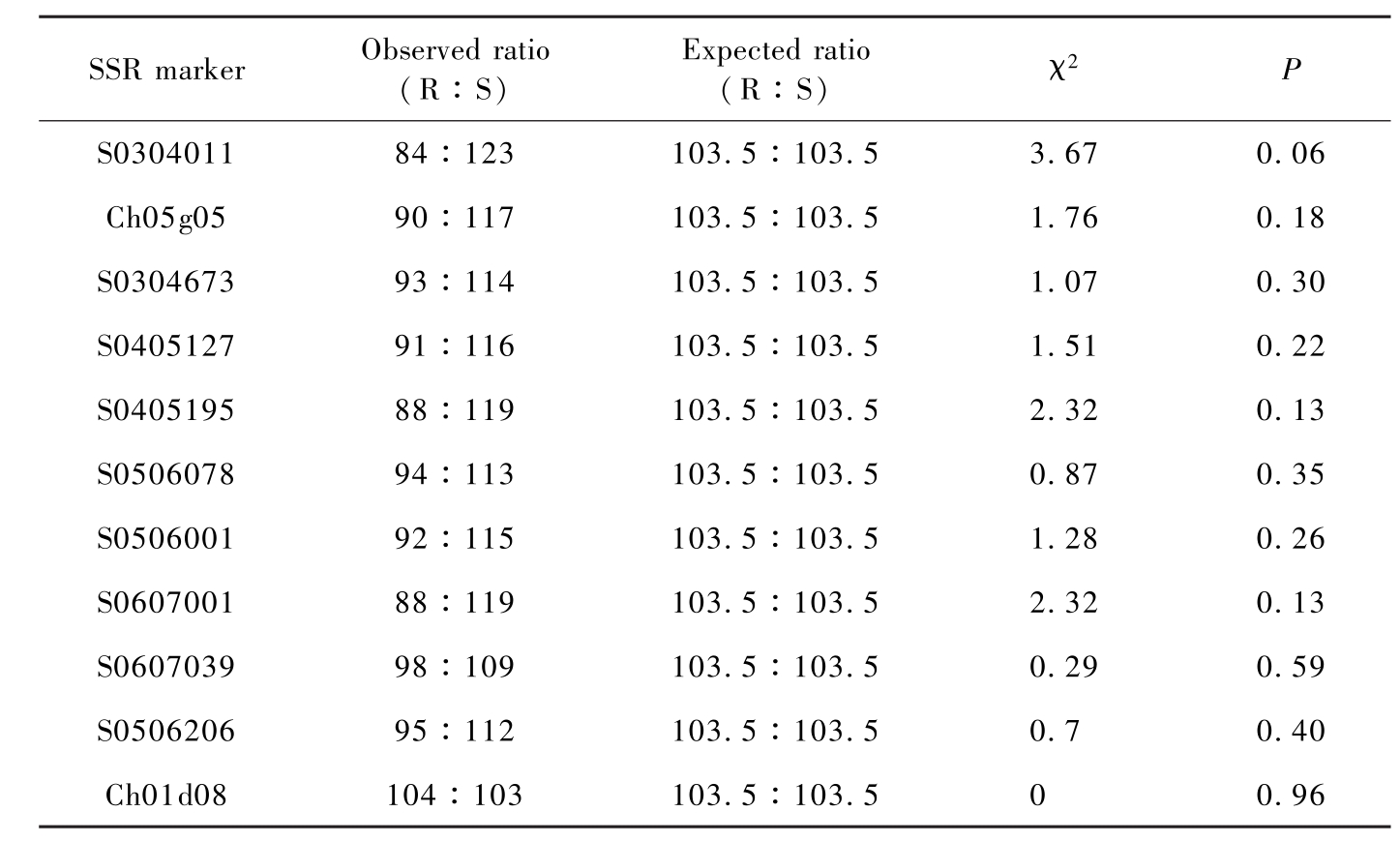
表3-2 SSR标记在207株 ‘金冠’ב富士’ F1 群体中的分离
三、遗传距离计算和抗性基因位点连锁图谱构建
将
为了确定抗性基因
四、SSR标记的测序分析
对S0304673 和 S0405127 进行测序分析。S0304673 标记能够在双亲中扩增出差异条带,而 S0405127 标记只在 ‘金冠’ 上扩增出一条带,所以对S0304673 标记在双亲中的扩增产物进行了测序,而只对S0405127标记在 “金冠” 上的扩增产物进行了测序 (附图 3-6、附图3-7)。
测序结果表明,SSR标记 S0304673 和 S0405127 的扩增片段大小分别为333 bp和330 bp。标记S0304673在 ‘富士’ 中的扩增片段比在 ‘金冠’ 中的扩增片段存在三处8~10 bp的碱基缺失,分别是 CT-CAGTGTGT、AGAGAAAG、CTTCTTACTT,另外还存在着一处两个碱基差异和六处单碱基差异。在 ‘金冠’ 中的扩增片段与参考基因组序列比对发现,有两处单碱基的差异,分别为 A/T和 G/A的碱基变化。标记S0405127在 ‘金冠’ 中的扩增片段与参考基因组序列比对发现,除在参考基因组中有两未知碱基以外,其余完全一致。本次测序确定了参考基因组序列的两处未知碱基分别为G和A。
第三节 讨论与小结
集团分离分析法 (BSA法) 是分子标记研究中的最经典的研究方法之一。其最大的贡献在于能够快速、有效地检测到与目的基因相连锁的分子标记,能够在连锁图谱中标记稀疏区或末端寻找到新的标记,并以此作为侧翼标记 (flanking marker),为继续寻找更紧密的连锁标记、构建高分辨率的连锁群、物理图谱和进行基因的图位克隆奠定基础 (廖毅,2009)。其原理简单、操作方便,而且克服了许多物种没有或者难以创建近等基因系的限制,被广泛地应用于作物育种中。同时必须注意到,物种基因组大小对标记与目标基因连锁距离是有影响的,一般来说基因组大,多态性少的物种,获得与目标基因紧密连锁标记的可能性也比较小。BSA法所能检测到的分子标记与目标基因的可信遗传距离一般在 15~25 cM,所以此法并不是在每一物种上都能获得所需要的目的标记 (Mackay and Caligari,2000)。DNA池的质量对BSA法的检测效率也有很大的影响。所以在实验过程中一定要注意,一是保证DNA 的纯度和浓度。杂质会影响紫外光的吸收率,高浓度的 DNA 溶解不均匀。因此混池时,尽可能使用高纯度 DNA,并适当稀释,否则会影响分池的精确性。二是避免 DNA 池污染。DNA 污染的原因有多方面,包括基因重组率、本身的表型效应、性状鉴定误差、DNA 混合误差、PCR 效率不均等。我们可以通过减少PCR 循环次数、减少混池单株数、构建多池、重复实验等方法来降低实验误差,否则这些误差将会导致多态性被覆盖而找不到目标标记。
随着分子生物学技术的快速发展,许多分子标记被成功的应用于控制农艺性状的重要基因的遗传定位及遗传图谱的构建。如 RAP D标记、RFLP 标记、SCAR 标记、CAPs 标记、SSR 标记、SNP 标记等。在这些标记中,SSR标记具有重复性好、可靠性高、共显性和适合自动化操作等优点,成为基因定位和遗传图谱构建的首选。而且,SSR标记广泛从布于整个基因组。据统计,大约有163426个 SSR 位点公
在前人的研究中,SSR标记 CH01d08 和 CH05g05 被定位在 ‘Fi-esta’בTotem’ 的遗传图谱中,遗传距离为 33.7 cM.而在本研究中,它们间的遗传距离为41.0 cM。这种类似的现象Paolo等 (2013)也报道过。他们在利用四个分离群体构建与柱型基因
理论上,标记在基因组上的遗传位置与物理位置应该是对应的。但是在本研究中发现,部分标记的遗传位置与物理位置并不是一一对应的。从附图3-4和附图3-5中可以看出,共有8个SSR标记的连锁图谱上的位置与在 ‘金冠’ 基因组序列中的物理位置是一致,另外3个标记,S0506078、S0405195 和 S0304011在遗传图谱上的位置与物理图谱上的位置不一致。这有可能是由于当前的苹果基因组重叠群序列产生装配错误,也有可能是苹果基因组中染色体结构的变异造成
在本研究中位于抗性基因
本研究中所利用的SSR标记,特别是新设计的276 对引物,有很大比例在亲本间能扩增出多态性条带,而在抗感池间无差异。虽然这部分标记与抗性基因
本研究首次开展了与抗炭疽病叶枯病基因









