基于WGR技术开发与苹果抗炭疽菌叶枯病基因相关联的SNP、Indel标记及抗病候选基因的鉴定
基于WGR技术开发与苹果抗炭疽菌叶枯病基因相关联的SNP、Indel标记及抗病候选基因的鉴定
SNP (单核苷酸多态性) 主要是指在基因组水平上由单个核苷酸的变异所引起的 DNA 序列多态性,包含单个碱基的转换、颠换等。InDel是指插入/缺失 (Insertion/Deletion) 基因组中小片段的插入和缺失序列,个体重测序中特指1~50 bp 的小片段插入和缺失。在生物的进化进程中,生物全基因组水平上积累了大量的微小变异,主要包括SNP 和InDel。这些微小变异的变异程度决定了物种之间在表型和生理结构等方面的差异,为新物种的形成提供了最原始的动力,是物种多样性的本质体现。
SNP 标记是基因组DNA序列中分布最广泛的一类标记,植物基因组中平均每数百bp就存在着一个SNP (Huang et al.,2010;Qi et al.,2013)。InDel标记也是一种重要的遗传标记,已被广泛应用于图位克隆、基因定位、动植物遗传多样性的鉴定、分子标记辅助育种等领域(Jander et al.,2002;Schnabel et al.,2005;王岩等,2009;王明军等,2010)。随着高通量测序技术的快速发展,利用全基因组重测序(whole genome re-sequencing,WGR) 技术结合混合分组分析法(bulked segregate analysis,BSA) 可高效检测出通过双亲杂交建立的后代群体中导致表型变异的 QTL和突变位点 (Abe et al.,2012;Takagi et al.,2013),并且可以识别大量的 SNP 和 InDel 位点,从而进行SNP 及InDel标记的开发,获得基因区间的SNP 及InDel标记。
本研究利用WGR技术及 BSA法相结合,获取与抗炭疽菌叶枯病
第一节 材料和方法
一、植物材料
用于全基因组重测序的材料为 ‘金冠’ ‘富士’ 及第三章中经过抗病鉴定的 ‘金冠’ב富士’ 的 F
用于qRT-PCR 分析的材料为 ‘富士’ 和 ‘金冠’ 带嫩叶的一年生健壮新梢各30 枝,用于室内人工离体接种。接种方法同第二章,分别于接种后0 h、12 h、24 h、36 h、48 h、60 h、72 h七个时间点采集叶片,液氮速冻后,用于提取RNA,检测基因表达。
二、DNA、RNA的提取,cDNA的合成及抗感池的构建
1.DNA的提取及纯度、浓度的测定
参考Doyle和Doyle (1987) 及 Cullings (1992) 提取基因组DNA的CTAB法,并加以改进,详见第三章。
利用1%琼脂糖凝胶电泳检测 DNA 的纯度和完整性。条带单一、清晰明亮、无降解、无污染的DNA样品作为合格样品用于后续研究。运用NanoPhotometer ? 分光光度计 (IMPLEN,CA,USA) 对DNA的纯度进行检测。DNA经过 Qubit ? DNA Assay Kit in Qubit ? 2.0 Flu-rometer (Life Technologies,CA,USA) 进行浓度的精确定量。将DNA
2.RNA的提取与检测
苹果嫩叶总RNA的提取方法如下。
(1) 将500 μl裂解液RLT Buffer、50 μl PLANTaid、5 μl β-巯基乙醇,依次加入灭菌的1.5 ml离心管中,混匀,待用。
(2) 将冻存于-70℃下的苹果嫩叶0.2 g置于预冷的研钵中,加少量液氮快速研磨成细粉状后转入上述待用的的离心管内 (样品容易褐化,动作要迅速,并保证液氮挥发完全)。
(3) 56℃温育5~10 min,期间不断剧烈震荡混匀,以保证充分裂解。
(4) 12000 r/min 离心10 min,将离心管缓慢从离心机中取出,将上清液转到新离心管中 (吸取上清液时一定要缓慢,尽量不要吸到底部沉淀)。
(5) 较精确估计上清液体积,加入 0.5 倍体积的无水乙醇,盖盖,上下颠倒混匀,此时可能会出现沉淀,但是不影响提取的过程。
(6) 将混合物 (每次上样应小于800 μl,可分两次加入) 加入吸附柱中,12000 r/min离心60 s,弃废液。
(7) 在吸附柱中加700 μl去蛋白液 RW1,室温下放置5 min (可稍延长时间,去除吸附柱上的蛋白污染),12000 r/min 离心30 s,弃废液。
(8) 在吸附柱中加入 500 μl 漂洗液 RW (加过无水乙醇),12000 r/min离心 30 s,弃废液。再加入 500 μl 漂洗液 RW,重复1遍。
(9) 将吸附柱放回空收集管中,12000 r/min空离心2 min,尽量除去吸附柱中残留的漂洗液。
(10) 将吸附柱放入一个 RNase free 离心管中,在吸附膜上加40 μl RNase-free water (最好事先在 70~90℃中水浴加热),室温放置2 min,12000 r/min 离心1 min,如需RNA浓度较高,可重复离心1次。
(11) 取4 μl RNA加入6×RNA Loading Buffer,用1%琼脂糖凝胶
(12) 将检测质量好的RNA放入-70℃下保存备用。
3.cDNA的合成
参照 PrimeScript RT reagent Kit with gDNA Eraser (Perfect Real Time) 试剂盒 (TaKaRa)。具体操作步骤如下 (全程冰上操作)。
(1) 基因组 DNA 的去除。在 0.2 ml 灭菌的离心管中依次加入1 μg 总 RNA,2 μl 5×gDNA Eraser Buffer,1 μl gDNA Eraser,用RNase-Free水补足10 μl,42℃,2 min (或者室温5 min);4℃。
(2) 反转录反应。在0.2 ml灭菌的离心管中依次加入10 μl步骤1的反应液,1 μl PrimerScript RT Enzyme Mix I,1 μl RT Primer Mix,4 μl 5×PrimerScript Buffer 2 (for Real Time),用 RNase Free 水补足20 μl。37℃,15 min;85℃,5 s;4℃。
(3) 质量检测。用 β-actin 基因的引物对反转录的 cDNA 进行PCR扩增,并用1%的琼脂糖电泳检测,选取质量好的样品在-20℃下保存备用。
4.抗感池的构建
将 ‘金冠’ב富士’ 的F
三、文库构建及库检
检验合格的4 份 DNA 样品每份取1.5 μg进行基因文库的构建。通过Covaris破碎机将DNA样品随机打断成长度为350 bp的片段。采用TruSeq Library Construction Kit 进行建库,严格使用说明书推荐的试剂和耗材。DNA 片段经末端修复、加 ployA 尾、加测序接头、纯化、PCR扩增等步骤完成整个文库制备。构建好的文库通过 illumina HiSeq
文库构建完成后,先使用 Qubit 2.0 进行初步定量,稀释文库至1 ng/μl,随后使用Agilent 2100对文库的插入片段 (insert size) 进行检测,符合预期后,使用Q-PCR 方法对文库的有效浓度进行准确定量 (文库有效浓度>2 nM),以保证文库质量。
四、上机测序
4个文库检验合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行 Illumina HiSeq TM PE 150 测序。围绕350 bp的片段进行双末端 125 bp 测序。表型差异的两个亲本测序深度为10 X,抗感池测序深度为20 X。
五、生物信息分析流程
信息分析的主要步骤如下 (附图4-2)。
步骤一:对下机得到的原始测序数据 (Raw data) 进行质控得到有效测序数据 (Clean data)。
步骤二:将Clean data比对到参考基因组上。
步骤三:根据比对结果,进行 SNP、InDel 的检测,分析 SNP、InDel的分布情况并进行注释。
步骤四:对子代SNP 频率差异进行分析。
步骤五:根据分析结果对目标性状区域进行定位。
步骤六:确定候选基因。
六、原始数据的获得与处理
测序得到的原始测序序列 (Sequenced Reads或者 raw reads),里面含有带接头的、低质量的 reads。为了保证信息分析质量,必须对raw reads过滤,得到有效测序序列 (clean reads),后续分析都基于clean reads。数据处理的步骤如下。
一是去除带接头 (adapter) 的reads pair。
二是当单端测序read中含有的N的含量超过该条read长度比例的10%时,需要去除此对paired reads。
三是当单端测序read中含有的低质量 (Q≤5) 碱基数超过该条read长度比例的 50%时,需要去除此对paired reads。
七、与参考序列的比对
有效测序数据通过 BWA (http://bio-bwa.sourceforge.net/) 软
八、SNP和InDel的检测及注释
人们采用GATK3.3软件 (McKenna et al.,2010) 的UnifiedGeno-typer模块进行多个样本 SNP 和 InDel的检测,使用 VariantFiltration进行过滤,SNP 的过滤参数为:cluster Window Size 4,filter Expression“QD<4.0 ‖ FS>60.0 ‖ MQ<40.0”,G filter “GQ<20”。InDel的过滤参数为:cluster Window Size 4,filter Expression “QD<4.0 ‖ FS>200.0 ‖ ReadPosRankSum<-20.0 ‖ InbreedingCoeff<-0.8”。利用ANNOVAR软件 (Wang et al.,2010) 对 SNP 和 InDel 检测结果进行注释。
九、SNP数据统计
SNP 频率的统计:SNP 频率是指所检测到的总的SNP 数与参考基因组序列的总长度的比值,是衡量一个物种变异程度和多态性的指标。转换颠换率的统计:分别统计发生转换和颠换的SNP 数目。
十、子代SNP频率差异分析
1.子代SNP频率计算
子代 SNP 的频率 (Takagi et al.,2013),即 SNP-index,是与SNP 位点的测序深度相关的参数,是指某个位点含有 SNP 的 reads数与测到该位点的总 reads数的比值。以参考基因组作为参照,分析计算两个子代在每个 SNP 位点的 SNP-index。SNP-index计算方法如附图4-3所示。若该参数为0,则代表所有测到的reads都来自作为参考基因组的亲本,即 ‘金冠’。参数为1则代表所有的reads都来自另一
为减少测序错误和比对错误造成的影响,对计算出 SNP-index后的亲本多态性位点进行过滤,过滤标准如下。
(1) 两个子代中 SNP-index 都小于0.3,并且 SNP 深度都小于7的位点,过滤掉。
(2) 一个子代SNP-index缺失的位点,过滤掉。
2.子代SNP频率差异分布
计算△ (SNP-index),即两个子代 SNP-index 作差:△ (SNP-index)=SNP-index2 (极端抗病性状)-SNP-index1 (极端感病性状)。为直观反映△ (SNP-index) 在染色体上的分布情况,对其在染色体上的分布进行作图。默认选择1 Mb 为窗口,1 kb 为步长。进行1000次置换检验,选取95%置信水平作为筛选的阈值。
十一、目标性状区域定位
为了不忽略掉微效QTL的影响,在全基因组范围内挑选两个子代在SNP-index差异显著的SNP 位点,即挑选子代2 (极端抗病性状) SNP-index大于0.7,且子代1 (极端感病性状) SNP-index小于0.3的位点,并与亲本 ‘富士’ 为纯合及亲本 ‘金冠’ 为杂合的位点取交集,做为候选的基因位点。提取ANNOVAR的注释结果,优先挑选使基因获得终止密码子的变异 (stop loss) 或者使基因失去终止密码子的变异 (stop gain) 或者非同义突变 (missense) 或可变剪接的位点(Splicing) 所在的基因作为候选基因。
十二、基因表达定量分析
从网站 https://www.rosaceae.org/gb/gbrowse/malus_x_domestica/下载候选基因的 mRNA,从基因编码区核苷酸序列的近3’端位置设计引物,PCR产物长度在150~250 bp,引物序列见表4-1,内参基因为β-actin基因。荧光定量 PCR 在罗氏 LightCycler 480 系统上进行,反应体系配制按 SYBR Green PCR Master Mix 说明书进行操
第二节 结果与分析
一、测序数据质量
Illumina HiSeqTM PE150 测序平台,四个样本共产生 47.115 G的测序原始数据 (Raw data),通过对原始数据中的接头序列,低质量碱基以及未测出的碱基 (用 N 表示) 进行过滤后,得到有效数据 (Clean data) 46.974 G,各样本的 Raw data 在 7774.247~17017.174 Mb,Clean date 在7751.990~16969.215 Mb,过滤后获得的有效数据比率在99.64%~99.72%,碱基错配率低于0.05%。测序质量 Q20 ≥ 95.08%、Q30 ≥ 88.97%,GC 含量在 39.04%~39.16%。这表明所有样本的数据量充足,测序质量很高,GC 分布正常,建库测序成功,保证了后续数据分析的准确性 (表4-1)。
样本原始数据/bp有效数据/bp有效率/%错误率/%Q20/%Q30/%GC含量/%富士7774246500775199010099.710.0395.4389.6339.16金冠7900566600787192650099.640.0395.5589.8839.05BR(基因抗池)170171739001696921590099.720.0395.4489.6239.04BS(基因感池)144234894001438116750099.710.0495.0888.9739.15
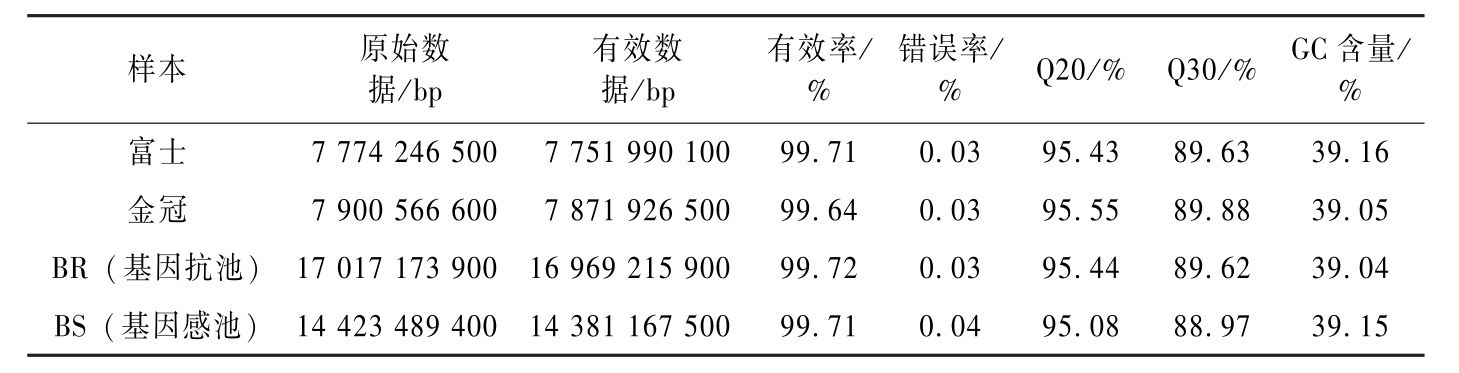
表4-1 测序数据质量概况
二、Reads与参考基因组比对情况统计
将四个样品的有效数据与已经发布的金冠苹果参考基因组序列(https://www.rosaceae.org/data) 进行比对,计算出比对到参考基因组 (参考基因组大小为609314018 bp) 上的 reads条数、测序深度及碱基覆盖率。结果表明,四个样品的比对率均在87%以上,两个亲本的测序深度达到15×以上,抗感池样品的测序深度达到 30×以上。在参考基因组中至少有1个碱基覆盖的位点占基因组的比例达到95%以上,至少有 4 个碱基覆盖的位点占基因组的比例 “富士” 为88.3%,“金冠” 为91.26%,其他两样品达到96%以上。这进一步的说明,本试验测序的数据质量很高,可用于后续的变异检测及相关分析 (表4-2)。
样本比对到reference上的reads条数①有效测序数据的reads总条数比对率/%②平均测序深度/x③参考基因组中至少有1个碱基覆盖的位点占基因组的百分比/%参考基因组至少有4个碱基覆盖的位点占基因组的百分比/%富士455966535167993488.2315.9595.9588.3金冠459536665247951087.5615.6798.0991.26BR(基因抗池)10045340711312810688.834.4298.896.67BS(基因感池)8962925610163794888.1830.8898.7896.44
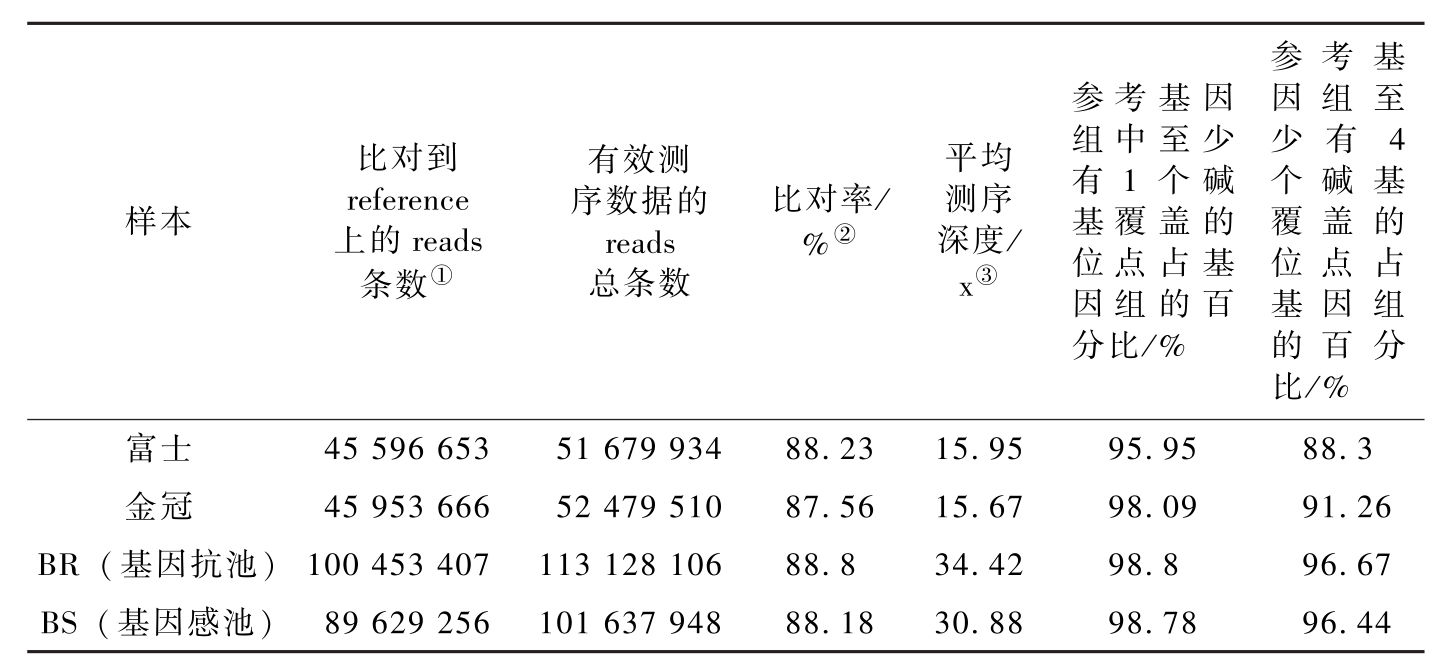
表4-2 测序深度及覆盖度统计
三、SNP频率和转换/颠换率计算
通过全基因组重测序技术共获得3399950个 SNP 位点,苹果参考基因组总长度为 609314018 bp,据此计算出 SNP 出现的频率为3399950/609314018=0.56%。转换 (T/C、C/T) 和颠换 (T/G、
四、SNP及InDel检测及注释
利用测序获得的有效数据,比对苹果参考基因组序列,经 ANN-OVAR软件分析共得到 SNP 位点 3399950个,InDel 位点 573040个,SNP 位点位于内含子上的465317个,位于外显子上的436309个,其中同义变异 210404个,非同义变异 220539个,InDel 位点位于内含子上的 108996个,位于外显子上的 19957个,其中插入或缺失3 或 3 的整数倍的碱基,不改变蛋白质的编码框的有 6928个。这表明绝大部分变异位于非编码区,或不会导致基因编码的改变,这有利于维持植物体正常的生长发育,保证品种特性的稳定遗传 (表4-3)。
类别SNP数量基因上游184156获得终止密码子4837失去终止密码子529外显子同义变异210404非同义变220539内含子465317剪接位点2196基因下游177312基因上游/基因下游12750基因间区2105122转换2265734颠换1134216转换/颠换/%1.997合计3399950

表4-3 SNP及InDel检测及注释
类别InDel数量基因上游51816获得终止密码子376失去终止密码子68Frameshiftdeletion①7330外显子Frameshiftinsertion5255Non-frameshiftdeletion②3843Non-frameshiftinsertion3085内含子108996剪接位点598基因下游44305基因上游/基因下游3896基因间区342800插入262868缺失310172合计573040

表4-3 SNP及InDel检测及注释(续)-1
五、子代SNP频率差异分布
△ (SNP-index)=SNP-index B (极端抗病性状)-SNP-index A (极端感病性状)。进行1000次置换检验,选取95%置信水平作为筛选的阈值。一般情况下,在苹果 F
六、目标性状区域定位
1.候选SNP 位点的筛选
在全基因组范围内挑选在两个子代池中 SNP-index 差异显著的SNP 位点,即挑选在抗池中 SNP-index 大于 0.7,在感池中 SNP-index小于0.3,△ (SNP-index) 大于 0.5 的 SNP 位点,并与亲本‘富士’ 为纯合,亲本 ‘金冠’ 为杂合的位点取交集,共得到258 个候选的多态性标记位点 (表4-4)。这258 个 SNP 位点中位于基因上游1 kb的19个,基因下游1 kb 的12 个,位于基因上游1 kb 区域,同时也在另一基因的下游1 kb区域的2 个,位于基因间区的173 个。位于外显子上非同义变异的13 个,同义变异7 个,位于内含子上的31个。其中位于第15条染色体上的有123 个,占整个候选 SNP 位点的47.7%。
类别SNP数量基因上游19获得终止密码子0失去终止密码子0外显子同义变异7非同义变13内含子31剪接位点1基因下游12基因上游/基因下游2基因间区173转换161颠换97转换/颠换/%1.659合计258

表4-4 候选多态性标记位点的注释
2.候选基因的确定
提取ANNOVAR的注释结果,优先挑选能引起基因获得终止密码子、失去终止密码子的变异,或者非同义突变,或者可变剪接的位点
基因ID类型染色体SNP位点参考基因组碱基型Ref突变碱基型AltMDP0000294705nonsynonymous24029182TCMDP0000311597upstream26560007TCMDP0000335197upstream238255166CTMDP0000481284nonsynonymous511687941TCMDP0000266783nonsynonymous516570950ACMDP0000223383upstream814935907CTMDP0000203531upstream918259400TGMDP0000265695upstream919790733GAMDP0000224187nonsynonymous1034991229ATMDP0000226279upstream1123158805AGMDP0000480608upstream123704417TGMDP0000504610upstream1229472316TCMDP0000292279upstream151396770CTMDP0000686092nonsynonymous153955717AGMDP0000686092nonsynonymous153955724AGMDP0000686092nonsynonymous153955738TAMDP0000205432nonsynonymous154094431TAMDP0000120033upstream154236293AGMDP0000864010nonsynonymous154257246GAMDP0000945764nonsynonymous154336468CAMDP0000149107splicing154970310CTMDP0000609131upstream156771435CAMDP0000169753upstream157074724ACMDP0000296372upstream157704309CT
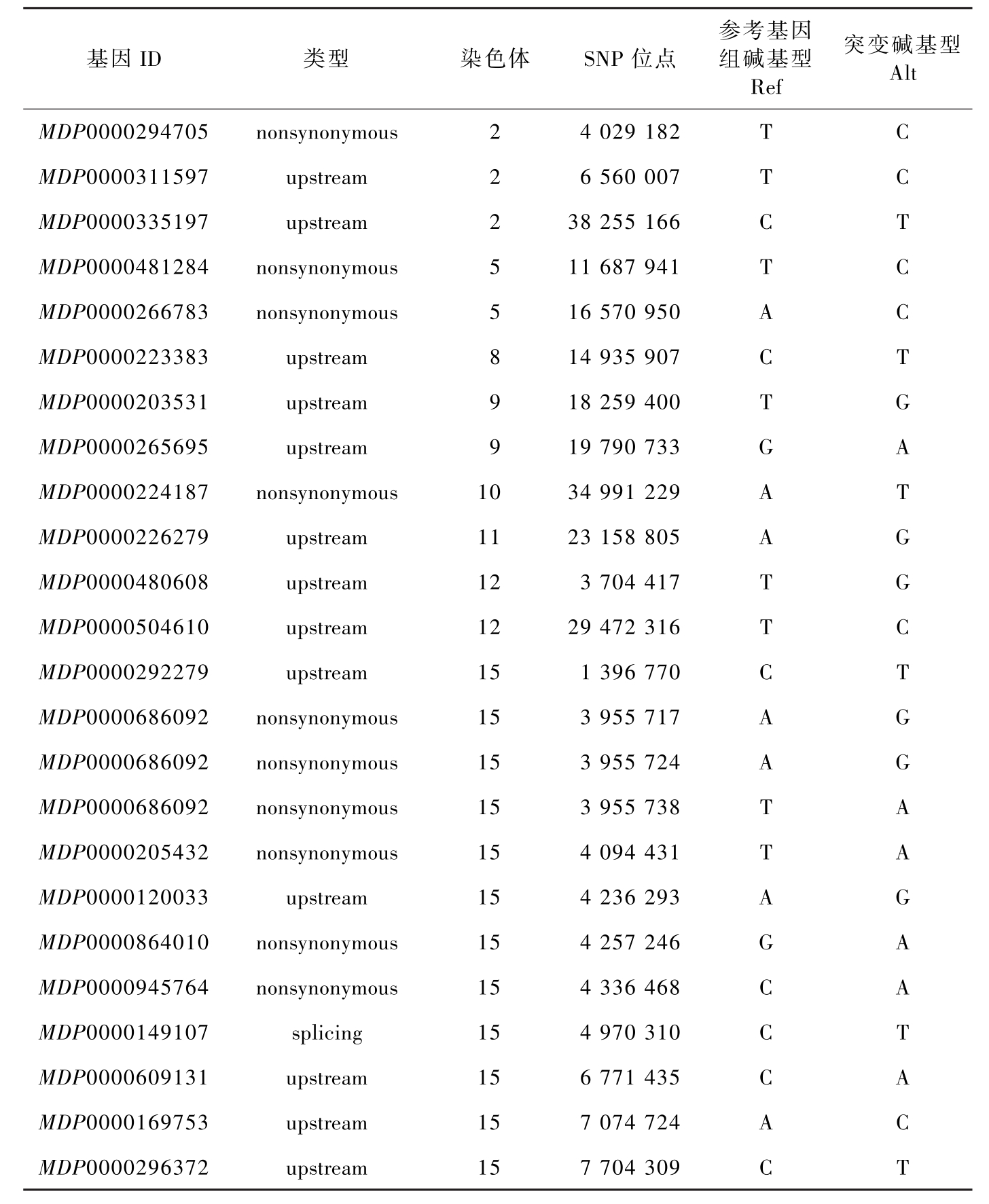
表4-5 候选基因ID
基因ID类型染色体SNP位点参考基因组碱基型Ref突变碱基型AltMDP0000199827upstream157710714TAMDP0000194508nonsynonymous157757532GAMDP0000320004upstream158084422ATMDP0000320004upstream158084636TCMDP0000320004upstream158084637ACMDP0000148808upstream158365795GTMDP0000133266upstream1533862275AGMDP0000157213nonsynonymous1549031042CGMDP0000169687nonsynonymous1552444208TC
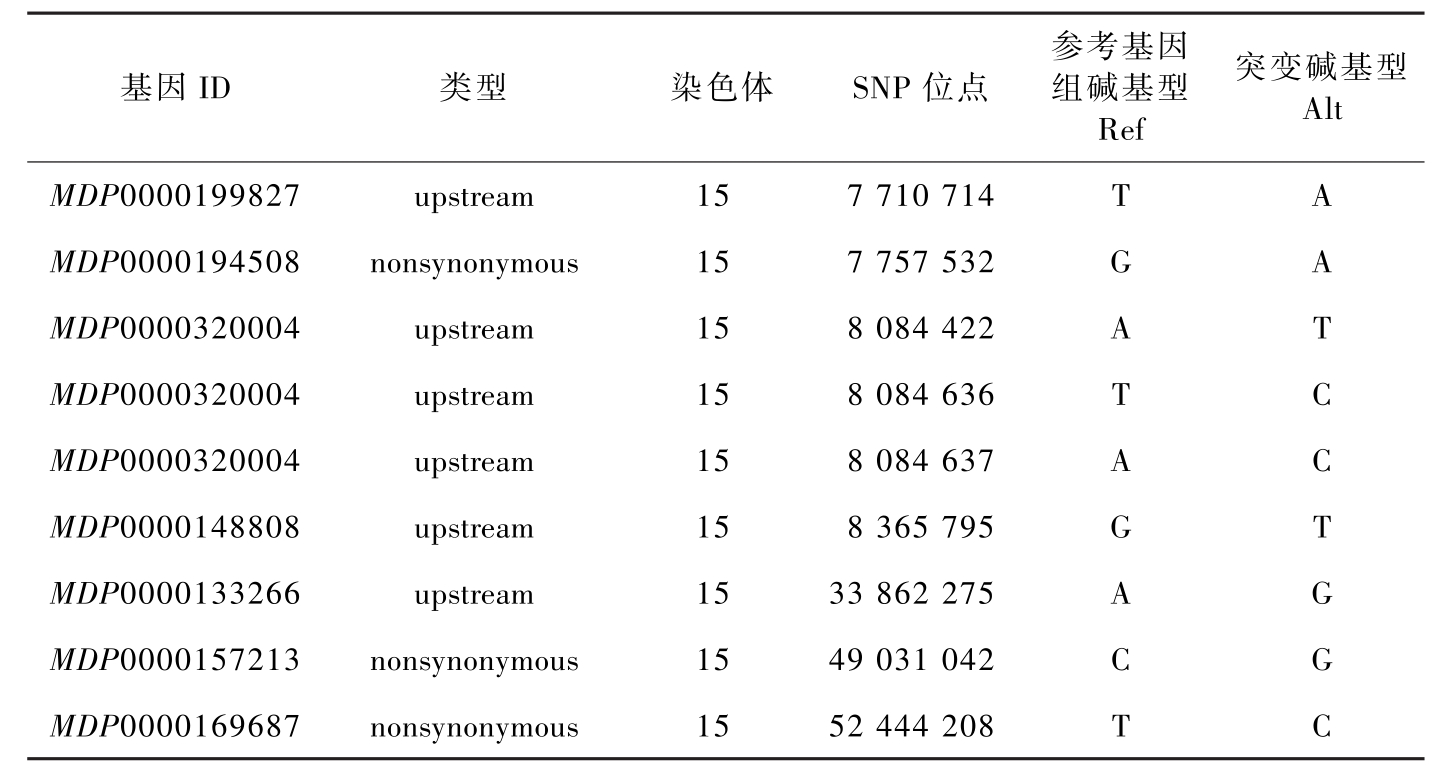
表4-5 候选基因ID(续)-1
结合 SSR标记定位及全基因组重测序中对△ (SNP-index) 值分析的结果,从这29 个候选基因中筛选出位于第15 条染色体3.9~4.9 Mb距离内的5个基因
七、候选基因的功能预测
1.GO功能富集分析
通过对5个候选基因进行 GO 功能富集分析发现,5 个候选基因中有3个功能未知。基因
基因ID基因功能注释MDP0000686092功能未知蛋白MDP0000205432功能未知蛋白质。MDP0000120033核酸绑定功能,锌离子结合分子功能,参与RNA剪切生物过程,调节基因产物的表达。具有锌指结构,CCHC型结构域,丝氨酸精氨酸富集剪接因子。MDP0000864010辅酶绑定功能,烟酰胺腺嘌呤二核苷酸(磷酸盐)NAD(P)绑定区域,NAD依赖差向异构酶/脱氢酶家族,可能与鼠李糖生物合成酶1有关。MDP0000945764功能未知蛋白

表4-6 抗炭疽菌叶枯病候选基因的功能注释
2.跨膜结构分析
利用TMHMM2.0软件对候选基因进行跨膜结构分析,基因
3.候选基因对炭疽叶枯病病原菌侵染的响应
引物名称引物序列MDP0000686092基因荧光定量PCR引物P1-FP1-RF:5′-CAGGCTGGAGCGAAGTTTA-3′R:5′-CCTTTCTGTGATGGCATTGTT-3′MDP0000205432基因荧光定量PCR引物P2-FP2-RF:5′-GAAAAGCCAGCACCAGAAAC-3′R:5′-CGTATTCGTGGGGTTATAGAGC-3′MDP0000120033基因荧光定量PCR引物P3-FP3-RF:5′-TCTGGGAATGCTGCTAAAACTC-3′R:5′-GCAGGGCAGTAGTGAAGCAC-3′MDP0000864010基因荧光定量PCR引物P4-FP4-RF:5′-CGTGACCAATCGCCTAATA-3′R:5′-GGACGTGAGTGCCGTAGAC-3
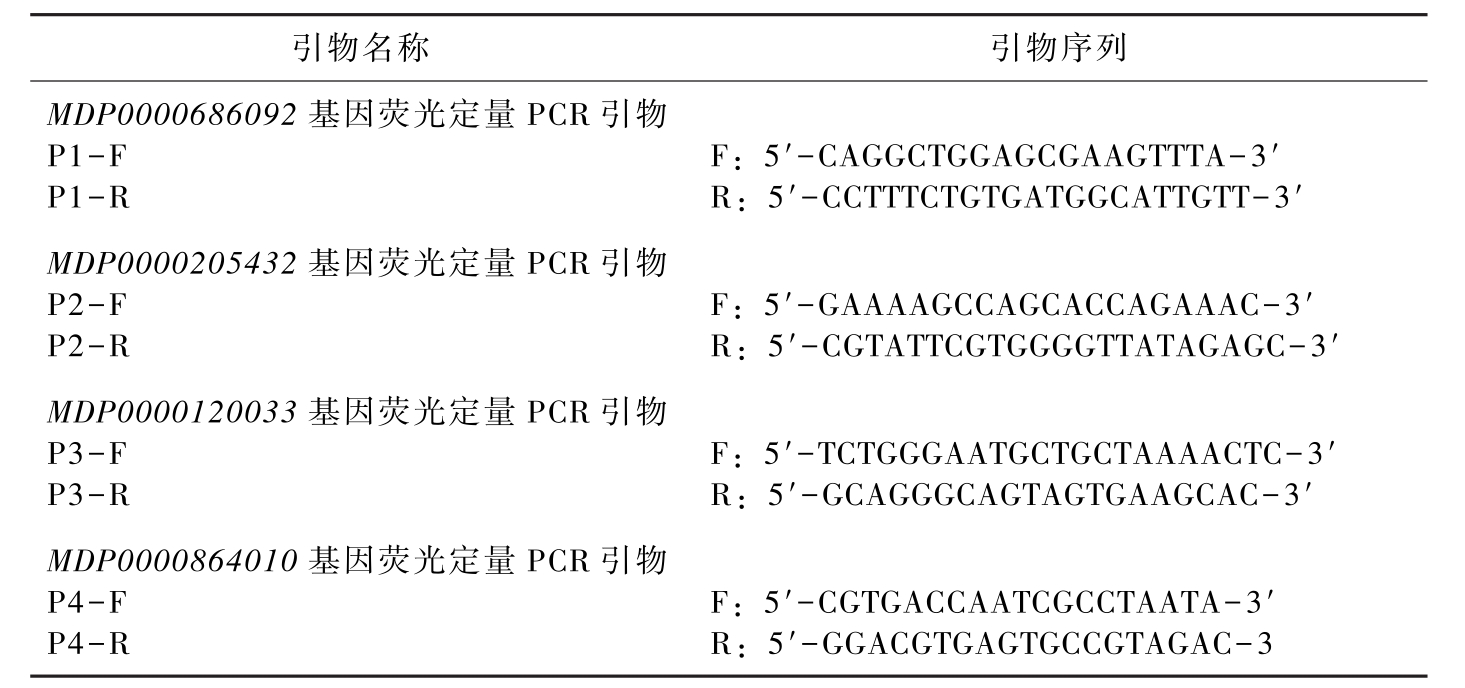
表4-7 荧光定量PCR引物
引物名称引物序列MDP0000945764基因荧光定量PCR引物P5-FP5-RF:5′-TTTGCCAGCCTGTCAGAAGT-3′R:5′-AGTTGTAGAGGGAGGAGGAAGA-3′β-actin基因荧光定量PCR引物P6-FP6-RF:5′-CACTGCTTCTATGACTGGTTTTGA-3′R:5′-CTGGCATATACTCTGGAGGCTT-3′
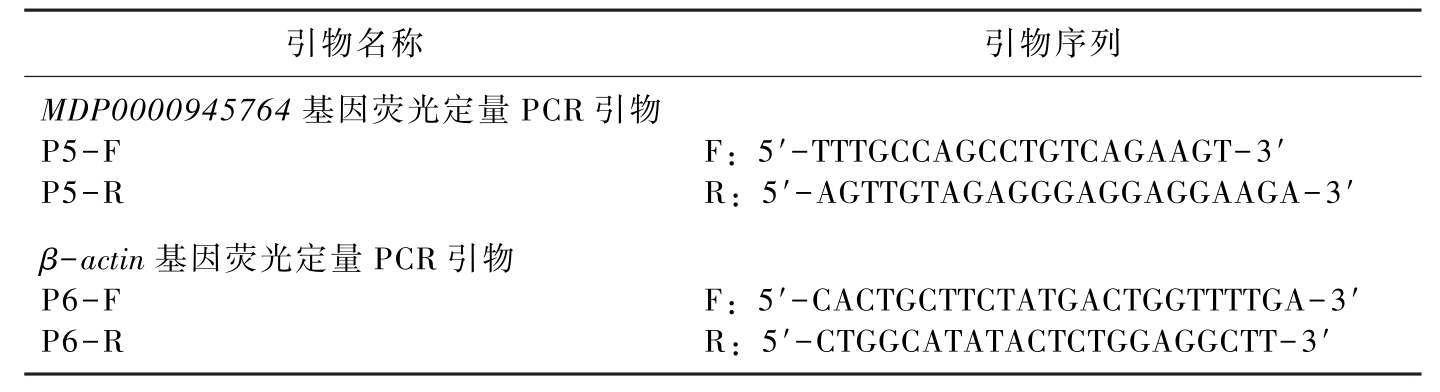
表4-7 荧光定量PCR引物(续)-1
对接种炭疽菌叶枯病病菌后 0 h、12 h、24 h、36 h、48 h、60 h、72 h的 ‘富士’ 和 ‘金冠’ 枝条进行采样,提取 RNA,反转录成cDNA后,对5个候选基因的表达进行实时荧光定量分析。结果显示,5个基因均不同程度响应病原菌侵染 (附图4-7、 表4-7)。除了基因
第三节 讨论与小结
SNP 标记的开发主要依赖于含有大量测序序列的数据库。通过全基因组测序,不仅可以获得大量的 SNP 标记,还可以了解到 SNP 标记在整个基因组中的分布情况,有利于我们进一步了解不同物种的生
SNP 变异从理论上来看主要包括转换、缺失、颠换和插入四种形式,但实际上发生的只有两种,即转换和颠换,二者之比为2∶1。并且SNP 在CG序列上出现的最为频繁,而且多是 C 转换为 T,主要原因为CG中的胞嘧啶常被甲基化,而后自发脱氨基形成胸腺嘧啶 T (Johnson and Told,2000;Mullikin et al.,2000)。从本实验中对 SNP转换/颠换频率统计结果来看,转换/颠换比为2.0,发生转换的频率为66.6%,发生颠换的频率为 33.4%,C/T 转换发生的频率最高为34.1%。这与前人研究的结论相符。
通过重测序技术可以获得海量SNP 标记,利用这些标记构建高密度遗传连锁图谱为不同群体的进化分析,不同性状基因的遗传定位,分子标记辅助育种等提供有效信息。‘M.27’בM.116’ 的高密度遗传图谱是由306个SSR标记和2272个 SNP 标记组成,图谱密度达到了每 0.5 cM 一个标记 (Antanaviciute et al.,2012)。Khan 等
SNP 广泛分布于基因组DNA中,且数量巨大。因为任何碱基均有可能发生变异,因此SNP 既有可能出现在基因的编码区,也有可能在基因的非编码区,或者两个基因之间的序列上。总的来说,位于基因内编码区的SNP 比较少,且该部分的 SNP 有可能会直接影响产物蛋白质的结构或基因表达水平,因此位于编码区 SNP 的研究更受关注。本试验对可能引起基因获得终止密码子、失去终止密码子、非同义突变、可变剪接的变异位点进行重点筛选,将其所在的基因作为候选基因。通过筛选共得到33个候选SNP 位点和29个候选基因。结合第三章SSR标记对炭疽叶枯病抗性基因的定位,最终锁定了 5 个候选基因:
锌指结构是一类在很多蛋白中存在的具有指状结构的模体,是具有识别特定碱基序列的一种转录因子结构。锌指的典型功能是作为互作的组件与核酸、蛋白质和小分子等多种物质相结合,参与多种细胞
可变剪切 (Alternative splicing,AS) 是重要的转录后调控机制,在动物中对其在不同的生理以及病程条件下的可变剪切的调控作用研究的较多 (Garcia-Blanco et al.,2004;Nilsen and Graveley,2010)。据报道在拟南芥中超过 42%的外显子基因存在可变剪切现象(Filichkin et al.,2010)。在植物可变剪切数据库中有很大比例的 AS参与逆境响应基因的表达,这进一步强调了可变剪切在逆境响应中的重要作用 (Wang and Brendel,2006;Duque,2011)。在生物胁迫中,有许多试验证明AS参与植物的抵抗和防御机制。例如,在一些 R 基因中如番茄的N 基因,拟南芥的
NAD依赖差向异构酶/脱氢酶家族是真菌病原体镰刀菌致病机制中的一个关键酶 (Srivastava et al.,2014)。细胞壁是植物细胞进行正常的生命活动不可缺少的重要组成部分。其不但在维持细胞形态、细胞间黏结、细胞壁的强度和调控细胞伸长等方面起着重要的作用,还参与了细胞的分化、抗病、细胞识别及信号传导等一系列生理、生化
基因的相对表达量分析也表明,五个基因都不同程度的响应了病菌的诱导,是与抗炭疽菌叶枯病相关的候选基因。但是其参与抗病的机理和途径仍不清楚,深入的研究将继续展开。









